To market and sell your medical device in Europe, you will need to comply with the Medical Device Regulation [MDR], and in the United States, with title 21 Part 820 of the Code of Federal Regulations [CFR]. HD’s process and systems follow the international ISO 13485 standard & the Code of Federal Regulations in the United States. According to 7.3.2 of ISO 13485, the international quality management system standard for medical devices, and FDA 21 CFR Part 820.30 (b), the quality system regulation for medical devices in the United States, plans for the design and development of a medical device shall be documented in a Design and Development Plan. The Design & Development Plan is a living document and shall be reviewed, updated, and approved as the project progresses.
ISO 13485 and the FDA’s CFR outline some loose guidelines for the development plan. The standard dictates that you need procedures in place and, to an extent, the requirements of those procedures. However, they don’t necessarily tell you what the procedure needs to be. Therefore, you’re required to write your own procedure. This is one of the reasons that there is often a lot of confusion surrounding the topic.
At HD, we have our own design & development procedure for medical device projects. We created this process internally utilising the experience of our team paired with our network of industry experts. Additionally, we have procedures in place to cover the requirements of ISO 14971, Risk Management for medical devices – another important factor to consider when planning the development of a medical device. We have also previously assisted pharmaceutical and healthcare clients with the development of their own set of audited systems and procedures as an essential part of developing and launching a medical device.
It’s important to note that a design development plan isn’t a ‘one size fits all’ activity neither for specific projects or companies.

Ash brings over 25 years of experience in leadership, board advisory roles and hands-on development within the health tech and medical device sectors. He has successfully guided numerous national and international companies through the complete product lifecycle from ideation to full-scale commercialisation and scale-up.

Early prototyping and user testing catch usability issues before costly mistakes, creating safer, smarter medical devices for users....

As we mark Love Your Lungs Week, we explore the latest trends shaping the inhalation industry. From advances in inhaler technology and sustainability initiatives to evolving patient needs and regulato...

Senior Design Development Engineer, Matt Holbrook shares an introduction to himself and how his first few months at Haughton Design have been....

As our newly appointed CEO enters his role, we ask him what he plans for the business in the future ahead....

Reflection from PDA Miniverse 2026: the big themes shaping medical devices, combination products and connected health....

As our Founder and Chairman retires, he reflects on building the company, the milestones along the way, and the future ahead....
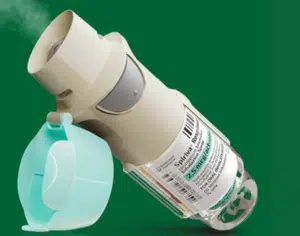
Explore inside a sustainable inhaler - do reusable designs really offer an eco-friendly solution, or is there more to consider than reusability....

HD CEO Ash Ghadar shares drug delivery development insights from PDA Vienna....

Discover how Haughton Design's diverse team leverages their extensive experience to drive innovation in device development. Learn about our expertise and unique insights that ensure successful project...

Explore how commercial constraints influence innovation in businesses. Discover whether budget limits, regulations, and market pressures drive creativity or stifle progress in today's competitive land...
